Die neue EU-Verordnung über Medizinprodukte – MDR
Mit dem Ziel, die Patientensicherheit zu verbessern, hat die Europäische Kommission die bestehenden Medizinprodukterichtlinien überarbeitet. Die neue Verordnung über Medizinprodukte (EU) 2017/745 (Medical Device Regulation, MDR) ist am 25. Mai 2017 in Kraft getreten, die Übergangsfrist endete am 26. Mai 2021. Sie ersetzt die bisherigen EU-Richtlinien 93/42/EWG und 90/385/EWG und muss nicht in nationales Recht umgesetzt werden, sondern ist unmittelbar in den Mitgliedstaaten gültig. Dies führt innerhalb der Europäischen Union zu einer Harmonisierung der Standards für Medizinprodukte.
Neben neuen Produkten müssen auch alle bereits auf dem Markt zugelassenen Produkte einem Zertifizierungsprozess gemäß MDR unterzogen werden. MDR-konforme Zertifikate können nur von Benannten Stellen vergeben werden, die bereits ein Neubenennungsverfahren durchlaufen haben. Dieser Vorgang ist derzeit noch nicht abgeschlossen (Stand September 2023).
Die wichtigsten Änderungen auf einen Blick
Die MDR bringt neue und in vielen Bereichen höhere Anforderungen für die Unternehmen und Überwachungseinrichtungen mit sich. Wichtige Änderungen sind:
- Für viele Produkte ändert sich die Klassifizierung: z. B. durch eine Höherklassifizierung von Software, für stoffliche Medizinprodukte, für wiederverwendbare chirurgisch-invasive Instrumente sowie für Produkte, die Nanomaterialien beinhalten.1)
- In der Risikoklasse I wurde die Untergruppe Ir (r für reusable) für wiederverwendbare chirurgische Instrumente eingeführt.2)
- Für aktive Produkte der Klasse IIb, die dem Körper Arzneimittel ab- oder zuführen und für Implantate der Klasse III ist ein Konsultationsverfahren im Zusammenhang mit der Klinischen Bewertung vorgesehen, bei dem Expertinnen und Experten in die Konformitätsbewertung einbezogen werden (Scrutiny-Verfahren).1)
- Die Anforderungen an die Klinische Bewertung sind gestiegen. Gleichzeitig wird der Verweis auf die klinischen Daten eines ähnlichen Produktes (Äquivalenzbetrachtung) stark erschwert und ist nach Meinung von Expertinnen und Experten schwer umsetzbar. Dadurch kommt es zu einem steigenden Bedarf an der Durchführung Klinischer Prüfungen – auch hier erhöht die MDR die Anforderungen.1)3)
- Die Anforderungen an das Qualitätsmanagementsystem, an die Technische Dokumentation und an die Überwachung nach dem Inverkehrbringen (Post-Market Surveillance) sind gestiegen.1)
- Mit der zeitlich nach Klasse gestaffelten Einführung einer neuen Produktidentifizierungsnummer (Unique Device Identification, UDI) ergeben sich darüber hinaus neue Kennzeichnungspflichten. Hierbei wird das Ziel einer besseren Nachverfolgbarkeit der Produkte verfolgt.3)
- Die Anforderungen an die Marktüberwachung sind gestiegen. Hersteller sind künftig verpflichtet, Daten über ihr Unternehmen, ihre Produkte sowie Informationen zu den UDIs in der EUDAMED-Datenbank zu hinterlegen, die als elektronisches Vigilanz- und Marktüberwachungssystem fungiert. Diese befindet sich weiterhin in der Umsetzungsphase. Bisher wurden die Module "Registrierung von Akteuren", "Produktkennung (UDI) und Registrierung" sowie "Benannte Stellen und Bescheinigungen" freigeschaltet (Stand April 2023).1)4)
- Jeder Hersteller muss intern mindestens eine für die Einhaltung der regulatorischen Vorschriften „Verantwortliche Person“ benennen, deren Aufgaben über die des oder der Sicherheitsbeauftragten hinausgehen. Ebenso steigen die Anforderungen bezüglich der Qualifikation und Erfahrung der „Verantwortlichen Person“.5) Laut "MDCG 2019-7 Guidance on Article 15 of the Medical Device Regulation (MDR) and in vitro Diagnostic Device Regulation (IVDR) regarding a 'person responsible for regulatory compliance' (PRRC)" kann diese Funktion in Kleinst- und Kleinunternehmen unter bestimmten Voraussetzungen auch extern besetzt werden.6)7)
- Große Veränderungen ergeben sich auch für die Zusammenarbeit zwischen Original Equipment Manufacturer (OEM) und Private Label Manufacturer (PLM). Nach derzeitigem Stand müsste der PLM vollen Zugriff auf die Technische Dokumentation des OEM erhalten, was Konfliktpotenzial im Bereich des geistigen Eigentums birgt.8)
Um Probleme und Fragen bei der Implementierung der MDR (EU) 2017/745 (Medical Device Regulation) sowie der neuen Verordnung über In-vitro-Diagnostika (EU) 2017/746 (In-vitro Diagnostic Medical Devices Regulation, IVDR) zu identifizieren, hat das Bundesministerium für Gesundheit den Nationalen Arbeitskreis zur Implementierung der neuen EU-Verordnungen über Medizinprodukte und In-vitro-Diagnostika (NAKI) gegründet. Die Ergebnisse werden auf der Website veröffentlicht.
Fristen
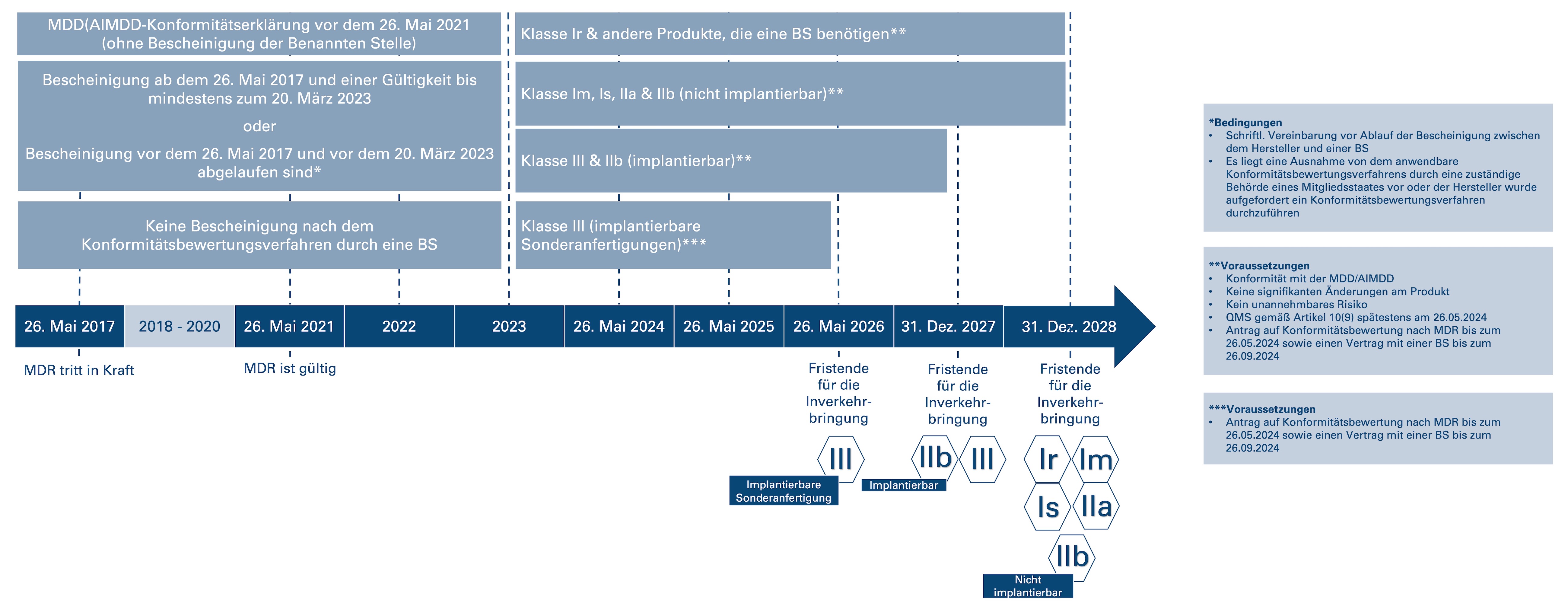
Am 15. März 2023 hat die Europäische Kommission die Anpassungsverordnung (EU) 2023/607 veröffentlicht, durch die die Übergangsbestimmungen für bestimmte Medizinprodukte angepasst wurden. Damit soll einem befürchteten Versorgungsengpass von Medizinprodukten in der Europäischen Union entgegengewirkt werden. Allerdings sind die Verlängerungen an bestimmte Bedingungen geknüpft. Die Europäische Kommission möchte sicherstellen, "dass die zusätzliche Zeit nur für Produkte gilt, die sicher sind, und für die die Hersteller bestimmte Schritte zum Übergang zur Einhaltung der Verordnung (EU) 2017/745 unternommen haben" (Abs. 6 (EU) 2023/607). Zusätzlich zu den verlängerten Übergangfristen für bestimmte Produkte wurde auch das Entfallen der Abverkaufsfristen beschlossen. Demnach ist die Bereitstellung auf dem Markt oder die Inbetriebnahme bestimmter Produkte auch nach Ablauf des Übergangszeitraums zulässig.
Bedingungen, an die die verlängerten Übergangsfristen geknüpft sind (Art. 1, Abs. 1 (EU) 2023/607):
- "Bescheinigungen, die von Benannten Stellen ab dem 25. Mai 2017 gemäß den Richtlinien 90/385/EWG und 93/42/EWG ausgestellt wurden, die am 26. Mai 2021 noch gültig waren, und die anschließend nicht zurückgezogen wurden, behalten ihre Gültigkeit bis zu dem Datum, das für die jeweilige Risikoklasse der Produkte festgelegt ist."
- "Bescheinigungen, die von Benannten Stellen […] ab dem 25. Mai 2017 [gemäß den Richtlinien 90/385/EWG und 93/42/EWG] ausgestellt wurden, am 26. Mai 2021 noch gültig waren und vor dem 20. März 2023 abgelaufen sind, gelten nur dann bis [zu den neu] festgelegten Zeitpunkten als gültig, wenn eine der folgende Bedingungen erfüllt ist:
- Vor Ablauf der Bescheinigung [müssen Hersteller und Benannte Stelle] eine schriftliche Vereinbarung […] über die Konformitätsbewertung des Produktes, für das die abgelaufene Bescheinigung gilt, oder eines Produktes, das dazu bestimmt ist, dieses Produkt zu ersetzen, unterzeichnet [haben].
- Eine zuständige Behörde eines Mitgliedstaats hat eine Ausnahme von dem anwendbaren Konformitätsbewertungsverfahren […] gewährt oder den Hersteller […] aufgefordert, das anzuwendende Konformitätsbewertungsverfahren durchzuführen."
Unter Berücksichtigung der oben genannten Bedingungen und den nachfolgenden Voraussetzungen (Art. 1, Abs. 1 (EU) 2023/607), dass
- die Produkte weiterhin der Richtlinie 90/385/EWG bzw. der Richtlinie 93/42/EWG entsprechen,
- keine signifikanten Änderungen am Produkt vorgenommen wurden,
- kein unannehmbares Risiko für die Gesundheit oder Sicherheit der Patientinnen und Patienten, Anwender oder anderer Personen besteht,
- der Hersteller bis spätestens am 26. Mai 2024 ein Qualtiätsmanagementsystem eingerichtet hat,
- bis spätestens am 26. Mai 2024 ein Antrag auf Konformitätsbewertung nach MDR sowie ein Vertrag mit einer Benannten Stelle bis zum 26. September 2024 unterzeichnet ist,
gelten für bestimmte Medizinprodukte folgende Übergangszeiträume (Art. 1, Abs. 1 (EU) 2023/607):
- Implantierbare Sonderanfertigungen der Klasse III dürfen bis zum 26. Mai 2026 ohne eine von einer Benannten Stelle nach dem Konformitätsbewertungsverfahren ausgestellten Bescheinigung in Verkehr gebracht werden, wenn der Hersteller bis spätestens 26. Mai 2024 einen Antrag bei einer Benannten Stelle gestellt hat und die schriftliche Vereinbarung bis spätestens 26. September 2024 unterzeichnet ist – dies gilt abweichend von Artikel 5 der (EU) 2017/745.
- Alle Produkte der Klasse III und implantierbare Produkte der Klasse IIb (ausgenommen Nahtmaterial, Klammern, Zahnfüllungen, Zahnspangen, Zahnkronen, Schrauben, Keile, Zahn- bzw. Knochenplatten, Drähte, Stifte, Klemmen und Verbindungsstücke) dürfen bis 31. Dezember 2027 in Verkehr gebracht werden.
- Klasse Im, Is, Klasse IIa und alle Klasse IIb-Produkte, die nicht implantierbar sind, dürfen bis zum 31. Dezember 2028 in Verkehr gebracht werden.
- Produkte der Klasse Ir oder andere Produkte, für die unter der (EU) 2017/745 eine Benannte Stelle für ein Konformitätsbewertungsverfahren erforderlich ist, dürfen bis 31. Dezember 2028 in Verkehr gebracht werden.
Nachfolgende Grafik veranschaulicht die momentan geltenden Übergangsfristen nach der Anpassungsverordnung (EU) 2023/607:
 Großansicht:
© BIOPRO Baden-Württemberg GmbH | Stand 04.2023
Großansicht:
© BIOPRO Baden-Württemberg GmbH | Stand 04.2023
Benannte Stellen
Nicht nur die Produkte selbst, auch die Zertifizierungsstellen, die die Produkte nach der neuen Verordnung zertifizieren, müssen sich erneut als Benannte Stelle bewerben und nach einer Prüfung neu benannt werden. Insgesamt gab es in der EU mehr als 50 Benannte Stellen mit der Befugnis, nach den EU-Richtlinien 93/42/EWG und 90/385/EWG zertifizieren zu dürfen.9) Im Zuge der neuen Verordnung haben 62 Benannte Stellen einen Antrag auf Benennung unter der MDR gestellt.10) 43 Benannte Stellen wurden MDR-konform neu benannt, davon zehn in Deutschland (Stand Januar 2024).11) Der aktuelle Stand kann in der NANDO-Datenbank abgefragt werden.
Wir machen ausdrücklich darauf aufmerksam, dass unser Webangebot lediglich dem unverbindlichen Informationszweck dient. Alle angebotenen Informationen sind ohne Gewähr und erheben keinen Anspruch auf Vollständigkeit und Richtigkeit. Eine Haftung für die Angaben sowie für deren Rechtsverbindlichkeit wird nicht übernommen.