BIOPRO Spezial: Die Klinische Prüfung im Blick
Klinische Prüfung von Medizinprodukten: Von der Planung bis zum Abschluss
Ab Mai 2021 ist die Verordnung (EU) 2017/745 über Medizinprodukte verpflichtend anzuwenden. Mit ihr steigt die Bedeutung der Klinischen Evidenz und somit der Klinischen Prüfung im Rahmen des CE-Konformitätsbewertungsverfahrens. Viele Hersteller müssen sich nun erstmals mit dem Thema der Klinischen Prüfung auseinandersetzen – Kenntnisse über den Ablauf einer Klinischen Prüfung für Medizinprodukte, von der Planungsphase bis hin zum Abschluss der Studie, sind für eine strukturierte Herangehensweise erforderlich.
Eine Klinische Prüfung ist laut Verordnung (EU) 2017/745 über Medizinprodukte (Medical Device Regulation, MDR) eine systematische Untersuchung, die zur Bewertung von Sicherheit oder Leistung eines Produkts durchgeführt wird und menschliche Prüfungsteilnehmer einbezieht.1
Bei Klinischen Prüfungen von Medizinprodukten ist zwischen verschiedenen Studientypen zu unterscheiden. Die Eingruppierung dieser Studientypen richtet sich im Allgemeinen nach folgenden Charakteristika:
- Regulierungsstatus
- Klinisches Entwicklungsstadium
- Studiendesign
- Belastung der Studienteilnehmer
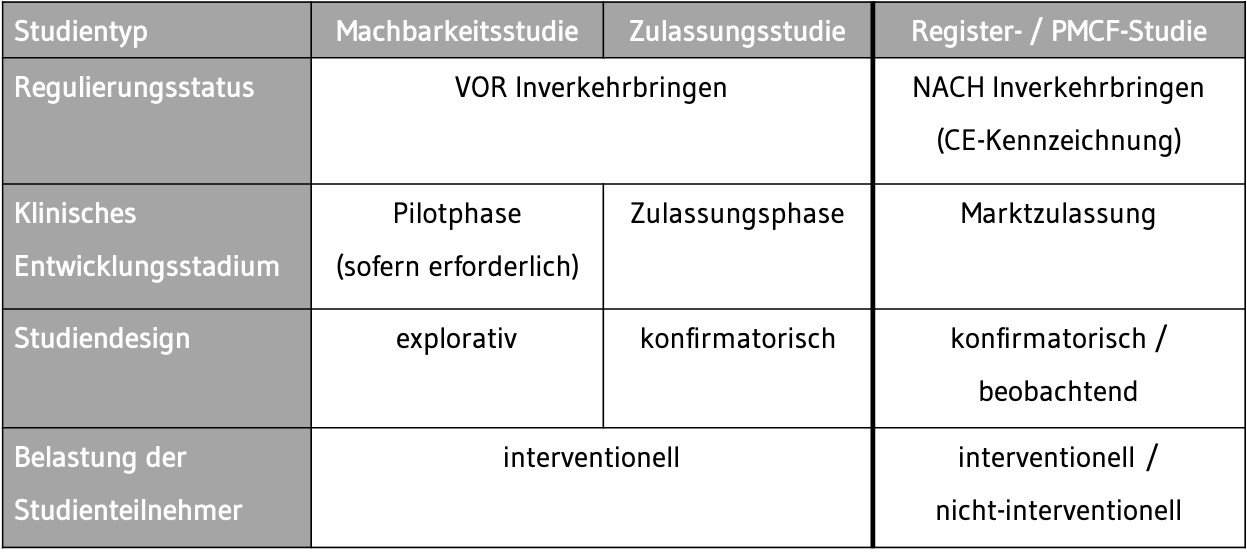 Tab. 1 In Anlehnung an die ISO 14155:2020-07: Klinische Prüfung von Medizinprodukten an Menschen – Gute Klinische Praxis (2)
Tab. 1 In Anlehnung an die ISO 14155:2020-07: Klinische Prüfung von Medizinprodukten an Menschen – Gute Klinische Praxis (2)Vor dem Inverkehrbringen des Medizinprodukts kann in einer Pilotphase eine erste Klinische Machbarkeitsstudie („First-in-Human“) an einer kleinen Gruppe von Studienteilnehmern (z. B. 10 – 30 Probanden oder Patienten) durchgeführt werden. Hier kann die Machbarkeit einer möglichen Klinischen Interventionsstudie überprüft und erforderliche Änderungen von Studienparametern für eine spätere Klinische Zulassungsstudie erkannt werden („explorativ“). In der Arzneimittelentwicklung werden die Klinischen Studien in Phasen von I bis IV eingeteilt. Diese Einteilung gibt es bei Klinischen Prüfungen von Medizinprodukten nicht.
In der Zulassungsphase werden die klinische Leistungsfähigkeit, Wirksamkeit und Sicherheit des Prüfprodukts bewertet. Sie dient der Überprüfung einer vorformulierten klinischen-wissenschaftlichen Fragestellung (Hypothese) von klinischen Endpunkten („konfirmatorisch“) und sollte eine ausreichend große Population an Studienteilnehmern aufweisen, um ein statistisch signifikantes Ergebnis zu erzielen.
Nach erfolgter Zulassung schließen sich häufig Klinische Prüfungen nach dem Inverkehrbringen an, sogenannte PMCF-Studien (Post-Market Clinical Follow-up Studies). Dabei wird die Anwendung des zugelassenen Medizinprodukts an einer breiteren Population (> 1000) von Anwendern und Patienten über einen längeren Zeitraum beobachtet („Beobachtungsstudie“). Im Gegensatz zu den Zulassungsstudien, die dem klinischen Prüfplan folgen, werden PMCF-Studien unter Alltagsbedingungen durchgeführt und zählen zu den nicht-interventionellen Studien.
Als Basis für eine strukturierte Herangehensweise und zur Durchführung einer Klinischen Prüfung nach ethischen Grundsätzen dient die ISO 14155.2, 3 Diese wurde zuletzt im Jahr 2020 grundlegend aktualisiert, um die gestiegenen Anforderungen durch die MDR widerzuspiegeln. Neben dem normativen und informativen Charakter der ISO 14155 müssen bei Studienvorhaben die gesetzlichen Verordnungen der MDR und die nationale Gesetzgebung beachtet werden. In Deutschland ist dies unter anderem das noch bis Mai 2021 geltende Gesetz über Medizinprodukte (MPG), das durch das Medizinprodukterecht-Durchführungsgesetz (MPDG) abgelöst wird.4, 5
Aufgrund der Komplexität ist es hilfreich, die Klinische Prüfung von der Planung bis zum Projektabschluss nach der zeitlichen Abfolge in verschiedene Projektphasen zu unterteilen:
1. Planungsphase
Die Planungsphase (Set-up Phase) einer Klinischen Prüfung kann in die administrative und logistische Planung und in die klinische-wissenschaftliche Planung unterteilt werden.
Die Herangehensweise und Umsetzung hängen dabei von mehreren Faktoren ab:
- Indikationsgebiet,
- Anwenderkreis,
- Risikoklasse des Medizinprodukts,
- Evidenzlücke (Restrisiken),
- Endpunkte in der Studie,
- Studientyp,
- Fallzahlen/Statistik,
- Zeitliche Planung/Budget/Ressourcen.
Ein zentraler Punkt ist die Formulierung der wissenschaftlich-klinischen Fragestellung, welche eine Studie am Menschen begründet und ethisch vertretbar macht. An dieser Stelle müssen vorhandene Evidenzlücken für das Medizinprodukt betrachtet und das Nutzen-Risiko-Verhältnis berücksichtigt werden. Ziel einer Klinischen Prüfung im Rahmen des Konformitätsbewertungsverfahrens und der Überwachung nach dem Inverkehrbringen ist es, die Erfüllung der grundlegenden Sicherheits- und Leistungsanforderungen des Medizinprodukts nachzuweisen.
Die klinische Fragestellung kann mit dem sogenannten PICO-Schema6 eingegrenzt werden und basiert auf den Evidenzlücken, welche im Plan für die Klinische Bewertung aufgezeigt wurden.
Das PICO-Akronym steht dabei für folgende Bestandteile in der Fragestellung:
P - Patienten, Population oder Problem,
I - Intervention (auch Exposure, Determinanten, Prädiktoren),
C - Comparison, Control (Kontrolle),
O - Outcome (auch Endpunkt).
Beispiel einer klinisch relevanten Fragestellung und die Ableitung der Fragestellung nach dem PICO-Schema:
Wie ist der Stellenwert einer Stent-Implantation bei einer stabilen koronaren Herzkrankheit?
P = Patienten mit stabiler koronarer Herzkrankheit (KHK),
I = Leitliniengemäße medikamentöse Therapie,
C = Verglichen mit einer perkutanen koronaren Intervention (PCI) mit Stent-Implantation,
O = Wirksamkeit hinsichtlich der Vermeidung künftiger Herzinfarkte.
Mithilfe dieser Punkte kann sich dem Studiendesign genähert werden. Ein Biometriker kann bei der Erstellung des Studiendesigns, der Fallzahlplanung sowie der Methodik unterstützen. Bei der Erstellung des klinischen Prüfplans (auch Studienprotokoll, Clinical Investigation Plan, CIP) sollten interne und externe Stakeholder involviert werden: Entwickler des Produkts, Vertreter aus dem Bereich Regulatory-Affairs, Biometriker, klinische Experten, Studienkoordinatoren, klinische Monitore, Datenschutzbeauftragte, ggf. Ethikkommissionen, ggf. Behörden.
Das Team einer Klinischen Prüfung setzt sich aus verschiedenen Experten zusammen: 7
- Entwickler und Produktmanager,
- Biometriker (Formulierung der Fragestellung, Erstellung von Studiendesign, Fallzahlplanung, Festlegung statistischer Methodik),
- Klinische Experten (Ärzte, z. B. Unterstützung bei der Erstellung des Klinischen Prüfplans und spätere Durchführung der Studie als Prüfärzte),
- Assistenten (Study Nurses) und Studienkoordinatoren (medizinisches Fachpersonal im Klinikum oder der Arztpraxis für die administrativen Aufgaben und Datenerhebung der Prüfbögen (Case Report Forms, CRFs),
- Projektmanager beim Studien-Sponsor oder der CRO (z. B. Erstellung des Budgetplans und dessen Nachverfolgung),
- Datenmanager und Datenschutzbeauftragter (Erstellung von Erhebungsbögen und Datenbankkonzepten, Gewährleistung des Datenschutzes),
- Klinische Monitore (Anleitung und Überwachung der Datenerhebung und Studiendurchführung),
- Vertreter aus den Bereichen Regulatory -, Clinical -, Commercial Affairs,
- Ethikkommissionen, Bundesoberbehörde.
Ein Blick in ähnliche Studienvorhaben (z. B. über das Deutsche Register Klinischer Studien (DRKS) oder www.clinicaltrials.gov) und Protokollvorlagen kann die Erstellung dieses zentralen Dokuments beschleunigen und verbessern. Der Aufbau des klinischen Prüfplans ist in der ISO 14155:2020 beschrieben.
Aufbau eines klinischen Prüfplans nach ISO 14155:2020 (Anhang A):2
- Allgemeines:
Einführung,
Identitätsmerkmale des klinischen Prüfplans,
Sponsor,
Prüfleiter, koordinierender Prüfer und Prüforte,
Gesamtübersicht über die Klinische Prüfung - Bezeichnung und Beschreibung des Prüfproduktes
- Begründung für das Design der Klinischen Prüfung
- Risiken und Nutzen des Prüfprodukts und der Klinischen Prüfung
- Zielstellung und Hypothese der Klinischen Prüfung
- Design der Klinischen Prüfung:
Prüfprodukte und Vergleichsprodukte,
Versuchspersonen,
Verfahren,
Monitoringplan - Statistische Planung und Analyse
- Datenmanagement
- Änderungen vom klinischen Prüfplan
- Abweichungen vom klinischen Prüfplan
- Verwendungsnachweis des Produkts
- Übereinstimmungserklärung
- Verfahren zum Einholen der Einverständniserklärung
- Unerwünschte Ereignisse, unerwünschte Wirkung des Produkts und Produktmängel
- Population, die unter Druck gesetzt werden kann (falls zutreffend)
- Unterbrechung oder vorzeitige Beendigung der Klinischen Prüfung
- Veröffentlichungspolitik
Neben der klinisch-wissenschaftlichen Planung müssen auch administrative und logistische Themen angegangen werden. Hierzu zählen vor allem die Ressourcen- und Budgetplanung innerhalb des Unternehmens, welche für die Klinische Prüfung aufgebracht werden. Oftmals sind beim Hersteller, der die Klinische Prüfung durchführt und verantwortet (Studien-Sponsor), mehrere Abteilungen involviert, die von einem Projektmanager koordiniert werden.
Die Einreichung und Anmeldung der Studie bei der Ethikkommission und der Bundesoberbehörde (BfArM oder Paul-Ehrlich-Institut) wird meist von der Regulatory-Affairs-Abteilung koordiniert, die auch die Studienordner (Investigator Site Files) mit den wesentlichen Dokumenten nach ISO 14155 für die Studienzentren erstellt.
Eine weitere Aufgabe dieser Phase ist die Planung der Produktionskapazitäten und der Logistik für das Prüfprodukt. Somit soll später gewährleistet werden, dass das Medizinprodukt für die Studie in einer ausreichenden Menge zur Verfügung steht und die Vorgaben für das Labeling der Verpackung die Anforderungen erfüllt. Für diese Planung ist es somit wichtig, dass frühzeitig die Fallzahlen und die Anzahl der Studienzentren geplant werden.
2. Machbarkeitsanalyse
Nachdem die grundlegende klinische-wissenschaftliche Fragestellung geklärt ist, wird mit einer sogenannten Machbarkeitsanalyse (Feasibility Study) die Umsetzung des Studienvorhabens eingeleitet. Diese Analyse ist ein wichtiges Element der Klinischen Prüfung, da spätere Verzögerungen in der Ethikeinreichung, beim Studienstart und in der Patientenrekrutierung vermieden werden können. Studienerfahrene Partner wie Auftragsforschungsinstitute (Contract Research Organizations, CROs) oder Koordinierungszentren für Klinische Studien können bei der Analyse und Umsetzung unterstützen.
Folgende zentrale Faktoren werden bei der Machbarkeitsanalyse betrachtet, anhand derer auch eine Einschätzung des benötigten Budgets vorgenommen werden sollte:8
- Studienzentren:
Welche und wie viele Studienzentren sind geeignet?
Besteht bei den geeigneten Studienzentren Interesse, an der Studiendurchführung mitzuwirken? - Patientenrekrutierung:
Wie viele Patienten müssen gemäß Fallzahlplanung rekrutiert werden?
Ist die benötigte Patientenpopulation ausreichend vorhanden?
Sind die Ein- bzw. Ausschlusskriterien im klinischen Prüfplan in der Praxis zu erfüllen? - Studienbudget:
Ist das Studienbudget ausreichend?
Entstehen Kosten für die Patientenrekrutierung (z. B. Fahrkostenübernahme)?
Entstehen Untersuchungs- und Behandlungskosten?
Entstehen Kosten für die Patientennachverfolgung (z. B. durch Telefonate)? - Zeitplan:
Ist der Zeitplan einzuhalten? - Interne Ressourcen und Kapazitäten:
Können die benötigten Ressourcen (z. B. Studien-Monitoring, Logistik) intern besetzt werden, oder bedarf es externer Dienstleister? - Personal und Dienstleister:
Welches Personal wird benötigt (Prüfärzte, Studienkoordinatoren, Study Nurses, Monitore, weiteres Personal)?
Werden Dienstleister in Anspruch genommen (Projektmanagement, Studienkoordination, Monitoring, Statistik, Datenmanagement, Labor, Archiv, weitere Dienstleister)? - Infrastruktur und Logistik:
Wo werden die Studiendokumente aufbewahrt (sicherer Aufbewahrungsort)?
Welche Räumlichkeiten werden benötigt?
Welche Geräte werden benötigt?
Ist eine fristgerechte Lieferung der Prüfpräparate gewährleistet?
Ein wertvolles und pragmatisches Instrument für die Machbarkeitsanalyse ist eine Befragung von klinischen Experten (z. B. erfahrene Prüfärzte). Dabei erhält eine Auswahl von klinischen Experten verschiedener Studienzentren einen noch nicht finalisierten Prüfplan bzw. eine Studiensynopse mit dem Studienablauf sowie den Ein- und Ausschlusskriterien. Die Expertenmeinungen werden dann gesammelt und ausgewertet. Somit ergibt sich eine Abschätzung der Machbarkeit (vorhandene Infrastruktur, ethische Fragestellungen) inklusive geschätzter Zeiträume für Rekrutierung einer ausreichenden Anzahl geeigneter Patienten.
Innerhalb der Machbarkeitsanalyse sollte auch die Strategie zur Einreichung und Anmeldung der Studie genauer betrachtet werden.
Innerhalb von Deutschland dürfen Klinische Prüfungen mit Klasse I und nicht-invasiven Produkten der Klasse IIa nur begonnen werden, wenn die zuständige Ethikkommission (EK) eine zustimmende Stellungnahme abgegeben hat, und sofern die zuständige Bundesoberbehörde nicht widersprochen hat (MPDG, §31 (1)). Bei allen anderen Produkten, wie zum Beispiel der Klasse III, darf eine Klinische Prüfung nur begonnen werden, wenn die zuständige Ethikkommission eine zustimmende Stellungnahme abgegeben hat, und sofern die zuständige Bundesoberbehörde eine Genehmigung erteilt hat (MPDG, §31 (2)).5
Die Stellungnahme der Ethikkommission und die Anzeigepflicht bei der Bundesoberbehörde sind nach MPDG nicht notwendig für Sonstige Klinische Prüfungen, wenn es sich um eine Studie mit einem CE-markierten Medizinprodukt innerhalb der Zweckbestimmung handelt, in der keine zusätzlichen invasiven oder andere belastenden Untersuchungen durchgeführt werden (MPDG, §47).5 Zu beachten ist jedoch, dass eine berufsrechtliche Beratung laut des Prüfarztes durch die Ethikkommission stattfinden sollte.
Die MDR unterscheidet zwischen der Klinischen Prüfung zum Nachweis der Konformität und der Sonstigen Klinischen Prüfung.10 Eine Sonstige Klinische Prüfung ist laut dem MPDG eine Klinische Prüfung, die nicht Teil eines systematischen und geplanten Prozesses zur Produktentwicklung oder der Produktbeobachtung ist, nicht mit dem Ziel durchgeführt wird, die Konformität eines Produktes mit den Anforderungen der MDR nachzuweisen, der Beantwortung wissenschaftlicher oder anderer Fragestellungen dient und außerhalb eines klinischen Entwicklungsplans nach Anhang XIV der MDR erfolgt.11
3. Anlaufphase
In der nun folgenden Anlaufphase (Study Start-up Phase) werden die wesentlichen Dokumente zur Klinischen Prüfung (Essential Clinical Investigation Documents) erstellt bzw. finalisiert und, sofern nötig, die zustimmende Stellungnahme der Ethikkommission sowie des BfArM (Bundesoberbehörde, BOB) eingeholt. Zu den wesentlichen Dokumenten, welche vor dem Beginn der Klinischen Prüfung vorliegen müssen, gehören zum Beispiel die Prüferbroschüre in dem unter anderem das Prüfprodukt beschrieben ist, der klinische Prüfplan, Versicherungsnachweise für die Studienteilnehmer, die Patienteninformation, die Einwilligungserklärung in die Studienteilnahme, die Einwilligung in die Verarbeitung personenbezogener Daten und die Prüfbögen (Case Report Forms, CRFs) für den Nachweis der zu erfassenden Studiendaten. Eine ausführliche Liste dieser Dokumente findet sich in der ISO 14155:2020 im Anhang E und im Anhang XV im Kapitel II der MDR.1, 2
Für die Einreichung bei der Ethikkommission und dem BfArM finden sich auf der Seite des Arbeitskreises Medizinischer Ethik-Kommissionen und auf der Seite des BfArM detaillierte Informationen, Checklisten sowie Vorlagen.12, 13
Sofern alle Dokumente, elektronischen Systeme (z. B. eCRF/EDC-System) und die Prüfprodukte zur Verfügung stehen und die Genehmigungen von Ethikkommission sowie Bundesoberbehörde (sofern erforderlich) erteilt wurden, kann mit der Studiendurchführung begonnen werden.
4. Durchführungsphase
In der nun beginnenden Durchführungsphase, die sich in die Initiierung und die anschließende Patientenrekrutierungsphase unterteilen lässt, werden die beteiligten Studienzentren in die Durchführung der Klinischen Prüfung auf Basis des klinischen Prüfplans eingewiesen. Dies erfolgt durch sogenannte Initiierungsbesuche (Site Initiation Visits, SIV), wobei hier die logistischen Anforderungen und die Qualifikation der Studienzentren durch einen klinischen Monitor (Clinical Research Associate, CRA) überprüft bzw. dokumentiert werden. Dies beinhaltet zum Beispiel die ordnungsgemäße Lagerung der Prüfprodukte.
Innerhalb eines Studienzentrums hat der Hauptprüfer (Principal Investigator) die Verantwortung für die Studie und wird von einem Studienteam aus Prüfern, Studienkoordinatoren und Assistenten (Study Nurses) unterstützt. Nach der Initiierung werden fortlaufend klinische Daten bei den Prüfungsteilnehmern gemäß des klinischen Prüfplans erhoben und in den (elektronischen) Prüfbogen dokumentiert. Die Einhaltung des klinischen Prüfplans wird kontinuierlich vom klinischen Monitor begleitet und überwacht.
Um die fortlaufende Qualität und Sicherheit der Patienten während der Studie zu gewährleisten, werden Monitoring-Visiten (Interim Monitoring Visits, IMVs) in festgelegten Intervallen an den Studienzentren durch den klinischen Monitor durchgeführt. Die Monitoring-Intervalle werden vom Sponsor festgelegt und im klinischen Prüfplan und im Monitoring-Plan festgehalten. Die Frequenz der Monitoring-Besuche richtet sich nach verschiedenen Faktoren, wie zum Beispiel der Rekrutierungsgeschwindigkeit der Prüfungsteilnehmer, Abweichungen vom Prüfplan und der Häufigkeit unerwünschter Ereignisse. Auch der Studientyp und die Klasse des Prüfpräparats spielen hierbei eine Rolle: Bei Zulassungsstudien mit einem Medizinprodukt der Klasse III werden Monitoring-Besuche häufiger durchgeführt als bei Registerstudien, bei denen eine Qualitätskontrolle durch Stichproben bzw. rein elektronisch auf Basis der Datenplausibilität vertretbar ist.
Ein besonderer Fokus des Monitorings liegt auf der Einhaltung der „Guten Klinischen Praxis“ (gemäß ISO 141552 und der Deklaration von Helsinki3). Dadurch wird gewährleistet, dass neben Datenplausibilität bzw. -verifizierung (Source Data Verification, SDV) auch überprüft wird, dass die Patienten aktiv in das Studienvorhaben eingewilligt haben und dies dokumentiert wurde. Andere essenzielle Punkte sind die Einhaltung des genehmigten Prüfplans durch die Prüfer sowie die Meldung von (schwerwiegenden) unerwünschten Ereignissen beim Einsatz des Prüfpräparates (Adverse Events, AEs/Serious Adverse Events, SAEs). Diese Aspekte sind auch bei der Überprüfung durch Audits bzw. Inspektionen durch den Sponsor und Behörden (z. B. BfArM und Regierungspräsidien) ein wesentlicher Bestandteil.
5. Studienabschluss
Nachdem die studienspezifische Behandlung bzw. Nachbeobachtung des letzten Studienteilnehmers abgeschlossen ist, kommt es zum Abschluss der Klinischen Prüfung (Close-out Phase). In dieser Phase werden die Studienzentren gegebenenfalls für ein letztes Monitoring besucht (Close-out Visit, COV), um noch offene Punkte in der Studiendatenbank zu überprüfen, bevor diese dann geschlossen wird. Nach dem letzten Besuch des letzten Prüfungsteilnehmers, teilt der Sponsor jedem Mitgliedstaat, in dem die Prüfung durchgeführt wurde, innerhalb von 15 Tagen das Ende der Studie mit (MDR Artikel 77 (3)).1 Hierbei gilt es zu beachten, dass die Klinische Prüfung auch vorzeitig beendet werden kann bzw. muss, wenn die Studien-Endpunkte vorzeitig erreicht wurden oder es sicherheitsrelevante Bedenken gibt.
In dieser abschließenden Phase werden alle studienrelevanten Ergebnisse in dem Bericht über die Klinische Prüfung (Clinical Investigation Report) zusammengefasst. Dieser Bericht wird innerhalb eines Jahres nach der regulären Beendigung der Klinischen Prüfung durch den Sponsor den Mitgliedstaaten über ein elektronisches System (EUDAMED) vorgelegt (MDR Artikel 77 (5)).1 Die Studienergebnisse sollten möglichst in einem wissenschaftlichen Artikel mit einem Peer-Review-Verfahren veröffentlicht werden, der dann in die Klinische Bewertung aufgenommen wird. Im letzten Schritt wird die Studiendokumentation nach den gesetzlichen Vorgaben durch den Sponsor archiviert. Die Aufbewahrungsfristen richten sich dabei nach der Art des Medizinprodukts und sind mindestens fünf bzw. 15 Jahre für implantierbare Produkte nach Beendigung der Klinischen Prüfung aufzubewahren (MPG §12/MDR Anhang XV Kap. III (3)).1, 4 Über diese Zeiträume muss die Dokumentation auch der zuständigen Behörde zur Verfügung stehen (MDR Anhang XV, Kapitel III/MPDG, §5 Absatz 2).1, 4
Wir machen ausdrücklich darauf aufmerksam, dass unser Webangebot lediglich dem unverbindlichen Informationszweck dient. Alle angebotenen Informationen sind ohne Gewähr und erheben keinen Anspruch auf Vollständigkeit und Richtigkeit. Eine Haftung für die Angaben sowie für deren Rechtsverbindlichkeit wird nicht übernommen.